
by Ian White, Soil Physicist, CSIRO Center for Environmental Mechanics, Canberra
ACT Australia,
Steven J. Zegelin, Soil Physicist, CSIRO Center for Environmental Mechanics,
Canberra ACT Australia, zegelin@python.enmech.csiro.au,
G. Clarke Topp, Soil Physicist, Agriculture and Agri-Food Canada, Ottawa, Canada,
toppc@ncccot2.agr.ca
Allison Fish, Research Student, The University of Sydney, Department of Soil Science,
Sydney, NSW, Australia.
Electric and magnetic fields propagating in wet porous media experience dielectric losses because of polarization and electrical conduction of the media. This work predicts the influence of dielectric losses on TDR-determinations of water content in porous materials and compares predictions with measurements. A three-phase effective medium model is modified to show how dielectric losses change the relation between apparent dielectric constant and water content without evoking bound water. This equation is tested using graphite-sand mixtures in which electrical conductivity varied systematically. Measured calibration relations were linear, as predicted by the model, but with slopes up to 250% larger than expected from the bulk phase properties of water. Independently determined low-frequency electrical conductivities of the samples were insufficient to account for the increases in slopes. Additions of electrolyte solution to samples confirmed that low-frequency conduction losses were not the principal cause of slope increases. The model suggests that the water-phase in these materials behaves as if its dielectric properties were substantially different from bulk water. It is suggested, by analogy with insulator-conductor composites, that high frequency conductivity losses may account for slope increases in porous materials.
The use of time domain reflectometry, TDR, to measure the frequency-dependent
complex relative permittivity, K*, of dielectric liquids (10) was
a major
innovation in
TDR technology. Its application to lossy dielectric soils to determine their apparent
dielectric constant, Ka, (21) and infer their volumetric water
content, ![]() , (4, 5, 21, 22)
was equally innovative, as well as practically important, and involved a considerable
leap of faith. (NOTE: A dielectric is described as lossy when it dissipates electromagnetic
energy during the polarization process or when it is slightly conducting.) This leap rests on the
premise that water in soil, or other porous materials,
is dielectrically identical to pure liquid water, despite differences in their partial Gibbs
free energy. In many porous materials, the water phase is both in intimate contact with
electrically charged surfaces and contains dissolved ionic species. Both cause dielectric
losses which can complicate the interpretation of TDR measurements of water content in
porous materials, especially heavy clay soils (1, 2,
7, 8, 12).
, (4, 5, 21, 22)
was equally innovative, as well as practically important, and involved a considerable
leap of faith. (NOTE: A dielectric is described as lossy when it dissipates electromagnetic
energy during the polarization process or when it is slightly conducting.) This leap rests on the
premise that water in soil, or other porous materials,
is dielectrically identical to pure liquid water, despite differences in their partial Gibbs
free energy. In many porous materials, the water phase is both in intimate contact with
electrically charged surfaces and contains dissolved ionic species. Both cause dielectric
losses which can complicate the interpretation of TDR measurements of water content in
porous materials, especially heavy clay soils (1, 2,
7, 8, 12).
Pore water in many porous materials does appear equivalent to bunk water (19).
Early TDR-measured Ka(![]() ) curves for many soils, particularly
light textured sands
and loams, cluster around a narrow region, suggesting a "universal" soil-independent
Ka(
) curves for many soils, particularly
light textured sands
and loams, cluster around a narrow region, suggesting a "universal" soil-independent
Ka(![]() ) relation (21). Subsequent work,
however, has shown that calibration
curves for
some low bulk density materials, such as organic soils, as well as media with
appreciable electrical conductivity and high specific surface, such as heavy clay soils
and anthracitic coals, do not obey the "universal" relation (7,
17, 26, 28). Some of
these departures may be due to inherent differences in the dielectric or magnetic
properties of the solid phase of the porous materials. Most differences, however, seem
attributable to the interaction of water with the porous material or the effect of
electrical conduction (8).
) relation (21). Subsequent work,
however, has shown that calibration
curves for
some low bulk density materials, such as organic soils, as well as media with
appreciable electrical conductivity and high specific surface, such as heavy clay soils
and anthracitic coals, do not obey the "universal" relation (7,
17, 26, 28). Some of
these departures may be due to inherent differences in the dielectric or magnetic
properties of the solid phase of the porous materials. Most differences, however, seem
attributable to the interaction of water with the porous material or the effect of
electrical conduction (8).
This work examines theoretical predictions of the impacts of these dielectric
losses, particularly electrical conduction, on TDR Ka(![]() )
calibration curves, and
compares these predictions with measurements.
)
calibration curves, and
compares these predictions with measurements.
PREDICTING THE INFLUENCE OF DIELECTRIC LOSSES
When electric and magnetic fields travel through wet porous materials their
energy can be dissipated by two major factors. The first is due to the fact that the
constituent molecules or dipolar species within a sample require a finite time, the
relaxation time, ![]() , to adjust to the changing field strength of the imposed electric
and
magnetic field (6). This polarization or relaxation process gives rise to a phase
lag
between the imposed field and the material's response to it. The phase lag is a
function of the angular frequency, co, of the imposed field. Because of this lag,
relative permittivity must be represented as a complex quantity, K*, with real
(in-phase), K'(
, to adjust to the changing field strength of the imposed electric
and
magnetic field (6). This polarization or relaxation process gives rise to a phase
lag
between the imposed field and the material's response to it. The phase lag is a
function of the angular frequency, co, of the imposed field. Because of this lag,
relative permittivity must be represented as a complex quantity, K*, with real
(in-phase), K'(![]() ), and imaginary (out-of-phase), K' (
), and imaginary (out-of-phase), K' (![]() ),
components (14).
),
components (14).
The second major factor causing dielectric losses in porous media is the
electrical conductivity of the media, ![]() . Conductivity can arise from both surface
conduction,
. Conductivity can arise from both surface
conduction, ![]() s, due to electric charges on the surface of the solids,
and from ionic
conductance,
s, due to electric charges on the surface of the solids,
and from ionic
conductance, ![]() w, caused by dissolved electrolytes in the water
phase. In sands and
loams
w, caused by dissolved electrolytes in the water
phase. In sands and
loams ![]() s is small, but in heavy clay soils it can be significant. The
contribution of
both loss factors to K* is given by (14)
s is small, but in heavy clay soils it can be significant. The
contribution of
both loss factors to K* is given by (14)
Here j = ![]() ,
, ![]() dc is the zero frequency, (dc)
electrical conductivity of the sample,
dc is the zero frequency, (dc)
electrical conductivity of the sample, ![]() 0 is
the permittivity of free space (8.85418x 10-12 F/m) and tan
0 is
the permittivity of free space (8.85418x 10-12 F/m) and tan![]() is the
loss tangent
defined as
is the
loss tangent
defined as
The loss tangent lumps together all dielectric losses. The functional dependencies
of K' and K' on ![]() for pure water are known (11) so the
frequency
dependence of
tan
for pure water are known (11) so the
frequency
dependence of
tan![]() for water can be calculated readily. In porous materials, water closely
associated with charged surfaces or ions is less mobile than in bulk water and is
called bound water. Bound water has a lower K* than pure water.
for water can be calculated readily. In porous materials, water closely
associated with charged surfaces or ions is less mobile than in bulk water and is
called bound water. Bound water has a lower K* than pure water.
Losses due to dipolar relaxation effects become important as the frequency of
the imposed signal increases to approach 1/![]() , but EQUATION 1
shows that dc conduction losses are more significant as frequency decreases. The hope has been
that TDR
measurements operate between these frequency extremes in a region where losses can
be ignored.
, but EQUATION 1
shows that dc conduction losses are more significant as frequency decreases. The hope has been
that TDR
measurements operate between these frequency extremes in a region where losses can
be ignored.
Electrical conductivity losses can sometimes cause total attenuation of the reflected signal. Attenuation in conducting porous materials becomes an increasing problem as the electrical conductivity or length of the TDR probe increases. Dalton and his colleagues (3) showed how attenuation can be used to estimate electrical conductivity.
The total down and back travel time, t, for a transverse electric and magnetic, TEM, wave propagating in an open-ended transmission line of length L imbedded in a medium with unit permeability and relative permittivity K*, is (24)
where c is the velocity of TEM waves in free space ( 2.997925x108 m s-1). TDR travel time measurements give the apparent dielectric constant, Ka as defined as (23)
From EQUATION 3 and EQUATION 4, it is clear that both in-phase and out-of-phase components contribute to the value of Ka
or in terms of the individual loss components
The frequency term in EQUATION 6 poses a problem for estimating Ka from the real and imaginary components of K* because TDR uses a band of frequencies rather than a single frequency. Heimovaara (13) concluded that Ka was the real part of the relative permittivity at the highest frequency of the TDR setup. However, he also concluded that Ka was influenced by the imaginary components as well, which is consistent with EQUATION 6.
It is expected from EQUATION 5 and EQUATION
6 that dielectric losses will cause Ka to
be greater than K' at the same frequency. How much greater depends on the
magnitude of tan![]() . For pure water the correction is much less than 1% at TDR
frequencies. Note that K', K' and
. For pure water the correction is much less than 1% at TDR
frequencies. Note that K', K' and ![]() dc in EQUATION 6 are
functions of
dc in EQUATION 6 are
functions of ![]() and
temperature.
and
temperature.
TDR measurements in sands to which electrolyte solutions has been added
suggest that Ka is, at best, a very weak function of conductivity of the soil solution.
Measured Ka(![]() ) for salt solutions appear identical to those
when water alone is added
(3, 23, 27). This may
be due to the fact that the dependence of dielectric constant on
the concentration of electrolytes in water is small (16).
) for salt solutions appear identical to those
when water alone is added
(3, 23, 27). This may
be due to the fact that the dependence of dielectric constant on
the concentration of electrolytes in water is small (16).
It follows from the above that the apparent dielectric constant, as measured by TDR, is a function of measurement frequency, temperature, electrical conductivity and the in- phase and out-of-phase components of relative permeability. These all interact. The dependence of Ka on water content is now examined.
Dependence of Apparent Dielectric Constant on Water Content
Early work on the relation between Ka and ![]() for soils was
pragmatic (21, 22).
TDR-measured Ka were calibrated against
for soils was
pragmatic (21, 22).
TDR-measured Ka were calibrated against ![]() determined by
oven drying at 105 degrees
Celsius. The
underlying notion was that oven drying is the accepted standard. In many environmental
applications it is mostly water which can be lost by oven drying that is of concern. The
pragmatic approach is totally agnostic about bound water.
determined by
oven drying at 105 degrees
Celsius. The
underlying notion was that oven drying is the accepted standard. In many environmental
applications it is mostly water which can be lost by oven drying that is of concern. The
pragmatic approach is totally agnostic about bound water.
Attempts to predict the bulk relative permittivity of a mixture from the volume fractions and relative permittivities of its components have been summarized recently (18, 19). A plethora of mixing laws have been adapted to porous materials despite the fact that most assume that one phase is discontinuous and the permittivity contrasts are small (19). Some models include contributions from four phases, solid, gas, free and bound water. When bound water is evoked, any mixture equation must be considered semi-empirical, since the volume fraction or the range of K for bound water are not known a priori. Both are usually treated as fitting parameters (7, 8).
Experimental evidence suggests that the relative permittivity of a wide range of porous materials follows the semi-empirical "refractive index" mixing model (1, 8, 15, 20, 25)
Here K*w and K*s are the relative
permittivities of the pore-water and solid phases, and ![]() b
and
b
and ![]() s are the dry bulk density and the particle density of the
material.
s are the dry bulk density and the particle density of the
material.
It is tempting to substitute TDR-measured Ka for the complex K in
EQUATION 7 and
indeed this has been done (25). This is only valid when dielectric losses are
negligible.
When losses are negligible, EQUATION 7 predicts that the square
root of Ka measured by
TDR in nonswelling materials should be a linear function of water content with slope of ![]() = 7.96, at 20°C. For sands, the linearity of measured
Ka1/2(
= 7.96, at 20°C. For sands, the linearity of measured
Ka1/2(![]() ) data is remarkably
good, although the slope is about 6% larger than the expected 7.96 (
26).
) data is remarkably
good, although the slope is about 6% larger than the expected 7.96 (
26).
In general, the real quantity Ka cannot be substituted for the complex quantity
K* in
EQUATION 7. Equating the real components of both sides of
EQUATION 7 and assuming
that tan![]() for the soil minerals is zero (8), one finds
for the soil minerals is zero (8), one finds
with Ka,w the apparent dielectric constant of the soil-water phase given by EQUATION 6.
EQUATION 8 predicts that the slope of the calibration line,
Ka1/2
versus ![]() , should be
linear with slope greater than Kw' by a factor {[l+(l+tan2
, should be
linear with slope greater than Kw' by a factor {[l+(l+tan2![]() w)/2}1/2, which depends on
the dielectric losses of the soil-water phase. Two key questions are: whether losses in the
soil-water solution are identical to those of bulk water with the same composition and
whether Kw' is that of bulk water; or whether the presence of soil minerals alters it.
Calculations of the losses for pure water show for the TDR frequency range that
(Ka,w-Kw')/Kw' < 0.01. Therefore, even for sands, the
observed increase in slope of 6% of the
calibration EQUATION 8 cannot be explained by
assuming that
water in the soil has the
same properties as pure water using this model.
w)/2}1/2, which depends on
the dielectric losses of the soil-water phase. Two key questions are: whether losses in the
soil-water solution are identical to those of bulk water with the same composition and
whether Kw' is that of bulk water; or whether the presence of soil minerals alters it.
Calculations of the losses for pure water show for the TDR frequency range that
(Ka,w-Kw')/Kw' < 0.01. Therefore, even for sands, the
observed increase in slope of 6% of the
calibration EQUATION 8 cannot be explained by
assuming that
water in the soil has the
same properties as pure water using this model.
It can be argued (18) that the dependence of the bulk electrical conductivity of a porous material on the conductivity of its constituents should follow a similar relation to that for relative permittivity. If so, we can write
where ![]() w is the conductivity of the pore water phase.
w is the conductivity of the pore water phase.
An examination will now be made of how well the suspiciously simple relation
between Ka and ![]() describes the TDR-measured relations for
materials whose bunk
conductivity was altered in a systematic fashion.
describes the TDR-measured relations for
materials whose bunk
conductivity was altered in a systematic fashion.
TESTING APPARENT DIELECTRIC CONSTANT RELATIONS
Porous Materials Used and Measurements
To change the bulk conductivity of a porous material systematically, powdered industrial graphite was mixed with Bungendore fine sand from which the clay fraction had been washed. Bungendore fine sand was used because its Ka versus 0 calibration curve is close to the theoretically expected curve (25) and is also in good agreement with the "universal" curve (21). The mixtures used ranged from 0% to 8% by weight graphite. Signal attenuation for samples above 8% graphite prevented accurate Ka measurement with uninsulated TDR probes. Water contents of the mixtures were changed by adding known quantities of distilled water to the mixtures. Duplicate calibration curves were determined for each graphite concentration. For two graphite concentrations, 0% and 3%, 0.005 M KCl solutions were added in place of distilled water as the water phase.
Soil-graphite mixtures with different water contents were packed into brass cylinders and Ka was measured using three-wire TDR probes (27) of length 0.148 m connected to Tektronics 1502B or 1502C cable testers. The tester was controlled and data was analyzed automatically and stored using a lap-top computer. Ten measurements were taken at each water content and the values averaged. The electrical conductivity of the graphite-sand-water mixtures was determined using an ac conductivity bridge (Radiometer, CDM3) at 120 KHz. The cell constant of the TDR probe for these conductivity measurements was determined using KCI solutions. All measurements were made at 20°C.
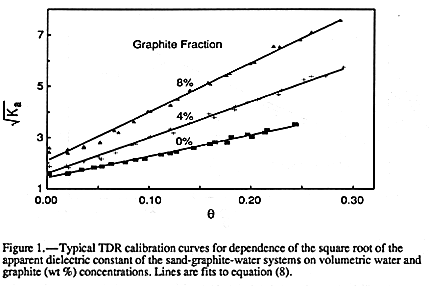
The influence of graphite concentration on the measured TDR calibration curve
between Ka and ![]() is shown in FIGURE 1
for three representative graphite concentrations,
0%, 4% and 8%. The duplicate measurements at each graphite concentration were not
significantly different and have been lumped together. Results for all graphite
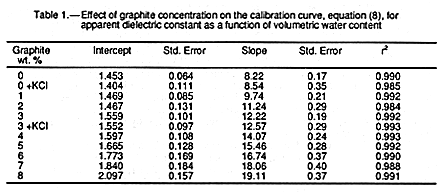
concentrations used are given in TABLE l.
is shown in FIGURE 1
for three representative graphite concentrations,
0%, 4% and 8%. The duplicate measurements at each graphite concentration were not
significantly different and have been lumped together. Results for all graphite
concentrations used are given in TABLE l.
Three main observations can be drawn from the results in
FIGURE 1 and TABLE 1. The
first is the remarkable linearity of the Ka1/2 against ![]() calibration curves for all added
graphite concentrations. The correlation coefficients listed in table 1 show an excellent fit
of the data to EQUATION 8. This linearity would appear consistent
with an hypothesis
that, for any particular graphite concentration, the soil-water phase behaves as if it has a
fixed apparent dielectric constant (Ka,w = constant) for the entire water content
range.
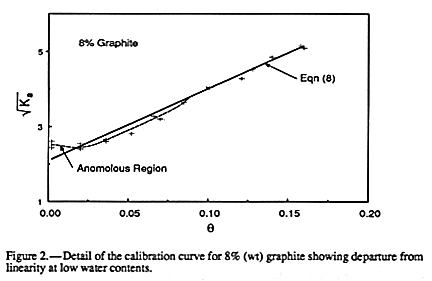
The second observation is that significant deviations from linearity do occur at water
contents less than about 0.05. This deviation is illustrated in
FIGURE 2 for the 8% graphite
data. Additions of water have either little impact on Ka up to
calibration curves for all added
graphite concentrations. The correlation coefficients listed in table 1 show an excellent fit
of the data to EQUATION 8. This linearity would appear consistent
with an hypothesis
that, for any particular graphite concentration, the soil-water phase behaves as if it has a
fixed apparent dielectric constant (Ka,w = constant) for the entire water content
range.
The second observation is that significant deviations from linearity do occur at water
contents less than about 0.05. This deviation is illustrated in
FIGURE 2 for the 8% graphite
data. Additions of water have either little impact on Ka up to ![]() of 0.05 or may well
decrease it. This seems to indicate formation of a bound water phase with a Ka less
than
that of the solid phase. Similar, although less pronounced, deviations occurred for all
graphite concentrations in this
of 0.05 or may well
decrease it. This seems to indicate formation of a bound water phase with a Ka less
than
that of the solid phase. Similar, although less pronounced, deviations occurred for all
graphite concentrations in this ![]() range. Note that EQUATION 8
cannot account for a
constant value of Ka at low
range. Note that EQUATION 8
cannot account for a
constant value of Ka at low ![]() .
.
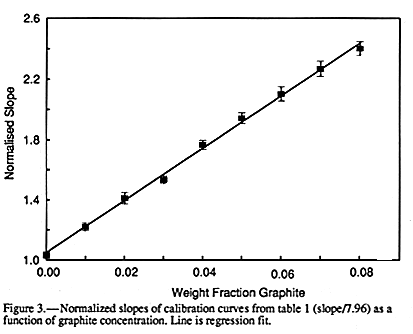
The third observation from FIGURE 1 and
TABLE 1 is the dramatic increase in slope of
the calibration curves with increasing graphite concentration. This is shown more clearly
in FIGURE 3 where the normalized slope of the calibration curves
is plotted against the
weight fraction of graphite. Here the slopes have been normalized relative to the slope
expected for pure water with no dielectric losses at 20#&176;C, ![]() = 7.96.
FIGURE 3 shows
that the slope can be increased by a factor of up to almost 250% and has an excellent
linear relationship with graphite concentration. The values of the intercepts (TABLE 1) appear independent of graphite concentration below
about 4%, after which they show a
significant increase. Deviations for the intercepts may be due to the above observation
that the measured Ka1/2 for small values of
= 7.96.
FIGURE 3 shows
that the slope can be increased by a factor of up to almost 250% and has an excellent
linear relationship with graphite concentration. The values of the intercepts (TABLE 1) appear independent of graphite concentration below
about 4%, after which they show a
significant increase. Deviations for the intercepts may be due to the above observation
that the measured Ka1/2 for small values of ![]() deviate significantly from the linear relation
dictated by EQUATION 8.
deviate significantly from the linear relation
dictated by EQUATION 8.
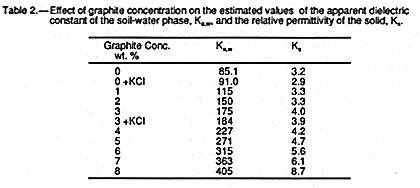
If the analysis presented in EQUATION 8 is valid then both the apparent dielectric constant of the soil-water phase (Ka,w) from the slopes in TABLE 1, and the relative permittivity of the solid phase (Ks) using the intercepts in TABLE 1, together with the mean particle and bulk densities of 2.62 and 1.48 t/m3, can be calculated. Values of Ka,w and Ks so calculated are given in TABLE 2.
Calculated values are listed in TABLE 2. The addition of graphite
alters the estimated
relative permittivity of the solid phase (Ks) significantly when the graphite
concentration
exceeds 3%. However, these changes are minor when compared with the apparent
dielectric constant of the water phase, Ka,w, whose values are spectacularly greater
than
the value of K0 for pure water (80.36 at 20#&176;C). Even Ka,w for
sand without added
graphite is significantly larger as is consistent with other measurements (25). If the
analysis underpinning EQUATION 8 is correct, then it seems that
the dielectric losses of
the soil-water phase are substantially increased by the presence of graphite in the sand.
This is unexpected since additions of conducting graphite (Ks![]()
![]() ) to sand (Ks
) to sand (Ks![]() 4.3)
ought to contribute to losses in the solid-phase component.
4.3)
ought to contribute to losses in the solid-phase component.
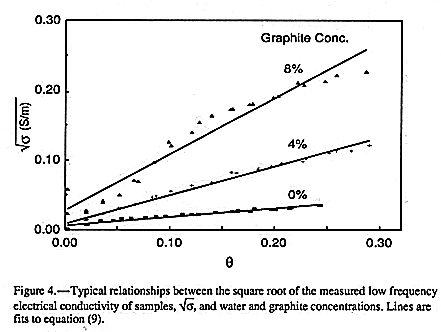
Graphite was added in these experiments to increase the electrical
conductivity of the bulk phase,![]() s. In EQUATION 9 both graphite
and quartz have
been lumped together into a single solid phase. FIGURE 4
shows typical relationships found between measured gin and H for three graphite
concentrations.
s. In EQUATION 9 both graphite
and quartz have
been lumped together into a single solid phase. FIGURE 4
shows typical relationships found between measured gin and H for three graphite
concentrations.
It is clear that the simple relationship, EQUATION 9, only holds in
an approximate
sense. Departures from linearity increase as the graphite concentration increases. At the
highest concentration (8% graphite) the dependence of ![]() on graphite
concentration
appears sigmoidal. Different functional dependences from that assumed in
EQUATION 9
seem to hold at low water contents. This may be consistent with the notion of the
presence of less mobile water.
on graphite
concentration
appears sigmoidal. Different functional dependences from that assumed in
EQUATION 9
seem to hold at low water contents. This may be consistent with the notion of the
presence of less mobile water.
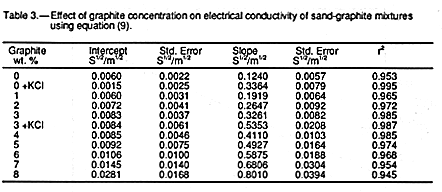
The values of the slopes, ![]() , and intercepts,
(
, and intercepts,
(![]() b/
b/![]() s
s![]() ,
found for each graphite
concentration fitted to EQUATION 9 together with standard errors
and the square of the
regression coefficient are given in TABLE 3. The regression
coefficients demonstrate that
the data are described only approximately by EQUATION 9.
FIGURE 5 shows the
dependence of
,
found for each graphite
concentration fitted to EQUATION 9 together with standard errors
and the square of the
regression coefficient are given in TABLE 3. The regression
coefficients demonstrate that
the data are described only approximately by EQUATION 9.
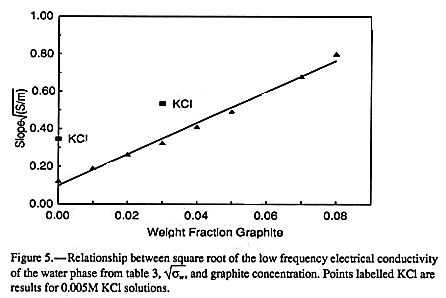
FIGURE 5 shows the
dependence of ![]() on graphite concentration. There it can be seen that,
despite the
approximate fit to EQUATION 9,
on graphite concentration. There it can be seen that,
despite the
approximate fit to EQUATION 9, ![]() appears to
be
an excellent linear
function of graphite
concentration. It is a simple matter to estimate the apparent electrical conductivities of
the solid (
appears to
be
an excellent linear
function of graphite
concentration. It is a simple matter to estimate the apparent electrical conductivities of
the solid (![]() s) and water (
s) and water (![]() w) phases
from the data in
TABLE 3.
w) phases
from the data in
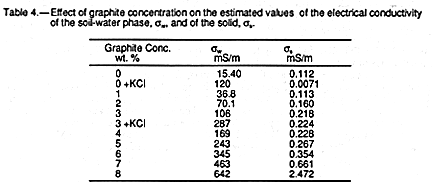
TABLE 3.
Calculated values of ![]() w and
w and ![]() s
listed in TABLE 4 show that it is only at the highest
graphite concentration, 8%, that the solid phase contributes significantly to the
conductivity of the sample. This is consistent with measurements in carbon black
composites (9). Both
TABLE 4 and FIGURE 4 demonstrate
that the major contribution to
electrical conductivity is in the soil-water phase at all graphite concentrations rather than
the solid phase (i.e. the dependence of slope on graphite concentration is greater than
that of the intercept).
s
listed in TABLE 4 show that it is only at the highest
graphite concentration, 8%, that the solid phase contributes significantly to the
conductivity of the sample. This is consistent with measurements in carbon black
composites (9). Both
TABLE 4 and FIGURE 4 demonstrate
that the major contribution to
electrical conductivity is in the soil-water phase at all graphite concentrations rather than
the solid phase (i.e. the dependence of slope on graphite concentration is greater than
that of the intercept).
Results for carbon black additions to two-phase, insulator-conductor composites show that these mixtures undergo a discontinuous transition from insulator to conductor at a particular additive concentration (9). This concentration is below that at which the conducting additive can form an interconnected network. The mechanism for conduction at this transition is believed to be governed by electron tunneling across insulating layers between particles or aggregates of the additive (9). The results here are consistent with the hypothesis that the water phase acts as a convenient conduit for conduction between graphite particles. If so, additions of small amounts of water should do little until a connecting network of water is formed. Once that network is formed, conduction should be independent of added water. Both seem consistent with the results in FIGURE 2, FIGURE 5, and TABLE 4.
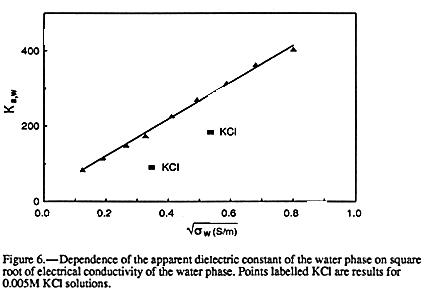
APPARENT DIELECTRIC CONSTANT AND BULK CONDUCTIVITY
If the sand-graphite-water systems studied here do behave as insulator-conductor
composites, it follows that there should be a strong correlation between the water-phase
conductivity and the apparent dielectric constant of the soil-water phase. This correlation
is shown in FIGURE 6 where Ka,w is plotted against
![]() and is
amazingly linear. A causal
relationship cannot be assumed here. Both quantities in
FIGURE 6 are determined by the
graphite concentration. In principle, EQUATION 5 can be used to
calculate the dependence
of the apparent dielectric constant of the soil-water phase, Ka,w, on its electrical
conductivity provided the loss tangent was known. It can be seen from
EQUATION 2 that in
order to calculate tan
and is
amazingly linear. A causal
relationship cannot be assumed here. Both quantities in
FIGURE 6 are determined by the
graphite concentration. In principle, EQUATION 5 can be used to
calculate the dependence
of the apparent dielectric constant of the soil-water phase, Ka,w, on its electrical
conductivity provided the loss tangent was known. It can be seen from
EQUATION 2 that in
order to calculate tan![]() , Kw' and Kw' must be
known and a representative value for the
measurement frequency must be identified. The first two quantities are unknown and the
third is a matter of some debate. If a representative value of the measurement frequency is
assumed to be 300 MHz and setting Kw' = 80.36 (pure water at 20°C)
Kw' = 0, the
contributions of
, Kw' and Kw' must be
known and a representative value for the
measurement frequency must be identified. The first two quantities are unknown and the
third is a matter of some debate. If a representative value of the measurement frequency is
assumed to be 300 MHz and setting Kw' = 80.36 (pure water at 20°C)
Kw' = 0, the
contributions of ![]() w to Ka,w can be estimated.
w to Ka,w can be estimated.
Substituting these values and the maximum ![]() w from TABLE 4
in EQUATION 2 for
w from TABLE 4
in EQUATION 2 for ![]() dc,
and using the resultant tano in EQUATION 1, it is found that the
maximum contribution to
Ka,w by conductivity alone amounts to only a 5.4% increase in apparent dielectric
constant above that for pure water. In order for dc electrical conductivity (
dc,
and using the resultant tano in EQUATION 1, it is found that the
maximum contribution to
Ka,w by conductivity alone amounts to only a 5.4% increase in apparent dielectric
constant above that for pure water. In order for dc electrical conductivity (![]() dc) alone to
account for the observed large increases in apparent dielectric constant, the representative
measurement frequency of the TDR would need to be assumed to be only 16 MHz. Since
the longest TDR pulse travel time measured in this work was of order 10-8 s
(i.e., a
characteristic frequency of l/10-8s = 100MHz), it seems highly unlikely that such a
low
representative frequency is reasonable.
dc) alone to
account for the observed large increases in apparent dielectric constant, the representative
measurement frequency of the TDR would need to be assumed to be only 16 MHz. Since
the longest TDR pulse travel time measured in this work was of order 10-8 s
(i.e., a
characteristic frequency of l/10-8s = 100MHz), it seems highly unlikely that such a
low
representative frequency is reasonable.
Given these assumptions, it appears from the data in TABLE 2 and TABLE 4 that the predominant contribution to the large apparent dielectric constants appears to be through the imaginary component, Kw' . It must also be added, however, that this component seems well correlated with the square root of the soil-water phase conductivity which is in turn related to the graphite concentration in the matrix. In insulator-conductor composites, high frequency losses appear due to ac conductivity which increases with frequency (12). A possible explanation of the large increases in apparent dielectric constant demonstrated in FIGURE 6 is that they are due to frequency conductivity caused by "tunneling" in the water-phase between graphite particles. Electrolyte was added to the water-phase to test this explanation.
Effects of Electrolytes in the Soil-Water Phase
Two experiments were performed in which 0.005M KCL was added to the dry soil in
place of distilled water for 0% and 3% graphite concentrations. The calibration curves for
![]() found in these experiments do show some systematic curvature however
they
agreed
well with the distilled water data. Values for the slope and intercept of
EQUATION 8 for
added salt are also listed in TABLE 1.
Calculated values of Ka,w and Ks are shown in TABLE 2.
The values of Ka,w for KCl are larger than for water, but not significantly so. It
may be
concluded that the presence of added dilute electrolyte has a negligible impact on
measured Ka. This is consistent with available data (3,
23, 27).
found in these experiments do show some systematic curvature however
they
agreed
well with the distilled water data. Values for the slope and intercept of
EQUATION 8 for
added salt are also listed in TABLE 1.
Calculated values of Ka,w and Ks are shown in TABLE 2.
The values of Ka,w for KCl are larger than for water, but not significantly so. It
may be
concluded that the presence of added dilute electrolyte has a negligible impact on
measured Ka. This is consistent with available data (3,
23, 27).
The dependence of the square root of the conductivity for the 0.005 M KC1 results
as a function of water content, again showed a systematic departure from
EQUATION 9,
although the data fit the relation reasonably well. Values for the slope and intercept of
EQUATION 9 for added KCl are also presented in TABLE 3
and the corresponding values of
![]() w and
w and ![]() s are listed in TABLE 4.
s are listed in TABLE 4.
The KCl data are also plotted in FIGURE 5 and
FIGURE 6 for comparison with the water
values. It can be seen that the added KCl does not appear to change the slope of the
relationship between Ka,w and graphite concentration or ![]() ,
but
merely displaces it.
,
but
merely displaces it.
It is clear from these results that contribution of dissolved electrolytes in the soil-
water solution to conductivity is fundamentally different from that of the conducting
surfaces of the graphite in the soil. This is unfortunate. The hope had been that it may
have been possible to use TDR-measured electrical conductivity to correct TDR
determined Ka(![]() ) calibration curves and produce a universal
relationship.
) calibration curves and produce a universal
relationship.
A fundamental thesis in the application of TDR to water content determination in
porous material has been that the dielectric properties of the soil-water phase are similar
to those of bulk water. Increasing evidence has accumulated to show that this is not the
case, particularly in materials with high surface charge (7,
13, 17, 26). For these
materials, dielectric losses perturb the relationship between the TDR-measured apparent
dielectric constant, Ka, and the independently measured water content. This work
has
attempted to examine, in a systematic fashion, the impact of dielectric losses on the
relationship between Ka and ![]() for porous materials.
for porous materials.
It has been shown that the simple three-phase, semi-empirical mixing law predicts
that the slopes ![]() (
(![]() ) calibration curves should be greater
than that calculated using
the real part of the dielectric constant of bulk water because of dielectric losses.
Calculations for pure water reveal, however, that dielectric losses in pure water are
insufficient to account for the observed increase in slope of calibration curves, even for
sands. This leads to two possible conclusions. Either water in even the most simple
porous material-water systems has different dielectric loss properties to pure water or
else the analysis of what dielectric properties are measured using TDR is faulty.
) calibration curves should be greater
than that calculated using
the real part of the dielectric constant of bulk water because of dielectric losses.
Calculations for pure water reveal, however, that dielectric losses in pure water are
insufficient to account for the observed increase in slope of calibration curves, even for
sands. This leads to two possible conclusions. Either water in even the most simple
porous material-water systems has different dielectric loss properties to pure water or
else the analysis of what dielectric properties are measured using TDR is faulty.
Graphite-sand mixtures were used to explore the predictions of the model
concerning the impact (Jr bulk sample conduction on the ![]() (
(![]() ) calibration curves
determined with TDR. The results demonstrated the predicted linearity of the calibration
curves but slopes were up to 250% larger than those calculated from the properties of
pure water. The increase in slopes was a linear function of the concentration of graphite
in the sample. If it is assumed that the simple three phase model is correct then it
suggests that the soil-water phase in these mixtures has substantially different dielectric
properties than those of bulk water.
) calibration curves
determined with TDR. The results demonstrated the predicted linearity of the calibration
curves but slopes were up to 250% larger than those calculated from the properties of
pure water. The increase in slopes was a linear function of the concentration of graphite
in the sample. If it is assumed that the simple three phase model is correct then it
suggests that the soil-water phase in these mixtures has substantially different dielectric
properties than those of bulk water.
Despite the linearity of the calibration curves, significant and consistent deviations do occur at low water contents. These could be interpreted as the occurrence of bound water. These deviations are worth exploring.
It has been shown that independently measured electrical conductivity of samples at
low frequencies cannot explain the observed increases in Ka,w For any particular
graphite concentration the electrical conductivity appeared to follow a mixing law similar
to that for relative permittivity. As well, the square root of the electrical conductivity of
the soil-water phase showed a surprisingly good linear correlation with graphite
concentration. This lead to the observation that Ka,w was also a linear function of
![]() . If it is assumed that sand-graphite-water systems behave similarly to
insulator-conductor composites, then the conductivity results suggest that the water phase
may act as an electron "tunneling" conduit between the conducting particles. This
conduction conduit may give rise to high frequency ac conductivity losses which could
account for the observed magnitudes of Ka,w
. If it is assumed that sand-graphite-water systems behave similarly to
insulator-conductor composites, then the conductivity results suggest that the water phase
may act as an electron "tunneling" conduit between the conducting particles. This
conduction conduit may give rise to high frequency ac conductivity losses which could
account for the observed magnitudes of Ka,w
If these assumptions are correct then the results presented here suggest that the imaginary component of the relative permeability of the soil-water phase is dominant for the frequencies used in TDR measurements. Experiments in which KC1 was added to the soil-water solution showed negligible impact on the TDR calibration curves which is consistent with the notion that the dielectric losses are predominantly due to relaxation effects. Measurements in heavy clay soils, where it was possible to estimate K ', indicate that it plays a dominant role in determining the slopes of calibration curves.
Unfortunately, the results presented here suggest that it does not yet seem possible to use TDR-measured values of apparent dielectric constant and electrical conductivity to back-calculate a universal calibration relation. In a pragmatic sense this does not restrict the usefulness of TDR since the slopes of the calibration curves for nonswelling media are remarkably linear. This implies that calibration curves can be determined with relatively few measurements. This study has raised a number of issues which require resolution. TDR measurements alone at one temperature will not provide the complete answer. Instead, measurements in the frequency domain at a range of temperatures appear necessary.
This work was partially supported by NERDCC under grant no. 1198 and CSIRO Land and Water Care Program. We thank Dr Kevin O'Connor of US Bureau of Mines, Twin Cities Research Centre, for helpful editorial comments.
Last modified: 06-12-98
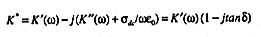{kind=link}
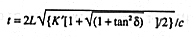{kind=link}
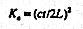{kind=link}
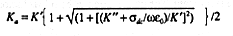{kind=link}
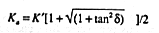{kind=link}
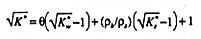{kind=link}
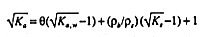{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
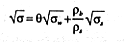{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
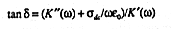{kind=link}